6月3日,我院生物物理团队黄胜友教授课题组在冷冻电镜的蛋白质结构建模领域取得重要进展,相关成果在线发表于《自然·通讯》(Nature Communications),题目为《基于同时的局部和非局部深度学习提升冷冻电镜密度图的可解释性》(Improvement of cryo-EM maps by simultaneous local and non-local deep learning)。华中科技大学为唯一通讯单位,我院博士生何佳铧为论文第一作者,博士生李涛为共同作者,黄胜友教授为通讯作者。

冷冻电镜已成为生物大分子复合物结构测定中最重要的技术之一,其目标是在三维重构得到的密度图上构建大分子复合物的原子模型。在建模过程中,密度图的质量至关重要。由于分子运动、结构异质性和不完美成像等固有因素的影响,三维重构得到的原始密度图往往存在信号对比度损失和局部分辨率不均一性等问题,无法立即用于准确的结构建模。因此,人们开发了各种算法,通过锐化或修改密度图来提高密度图的质量,这一步骤通常被称为冷冻电镜密度图后处理。然而,现有方法往往只实现了密度图质量在个别评价指标上的提升,而无法有效提升密度图在结构建模中的可解释性。
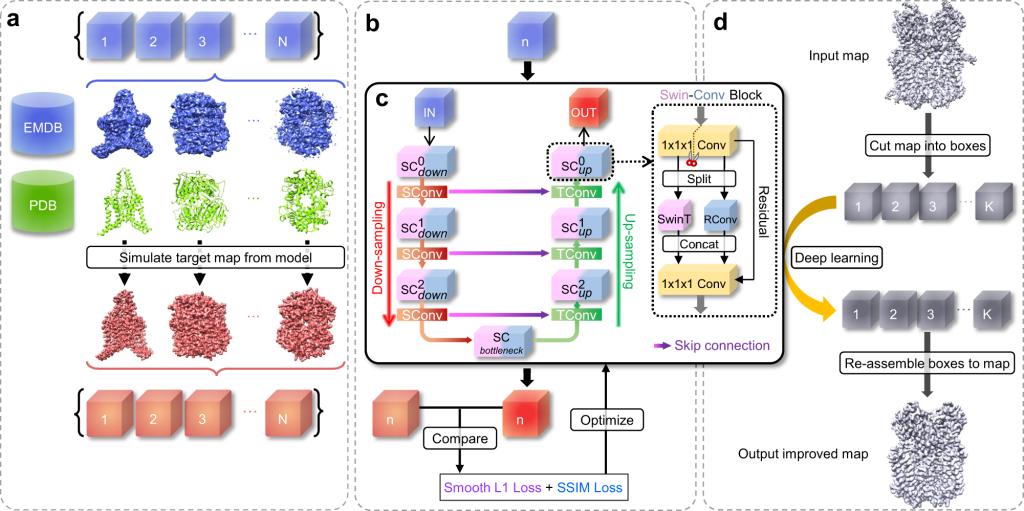
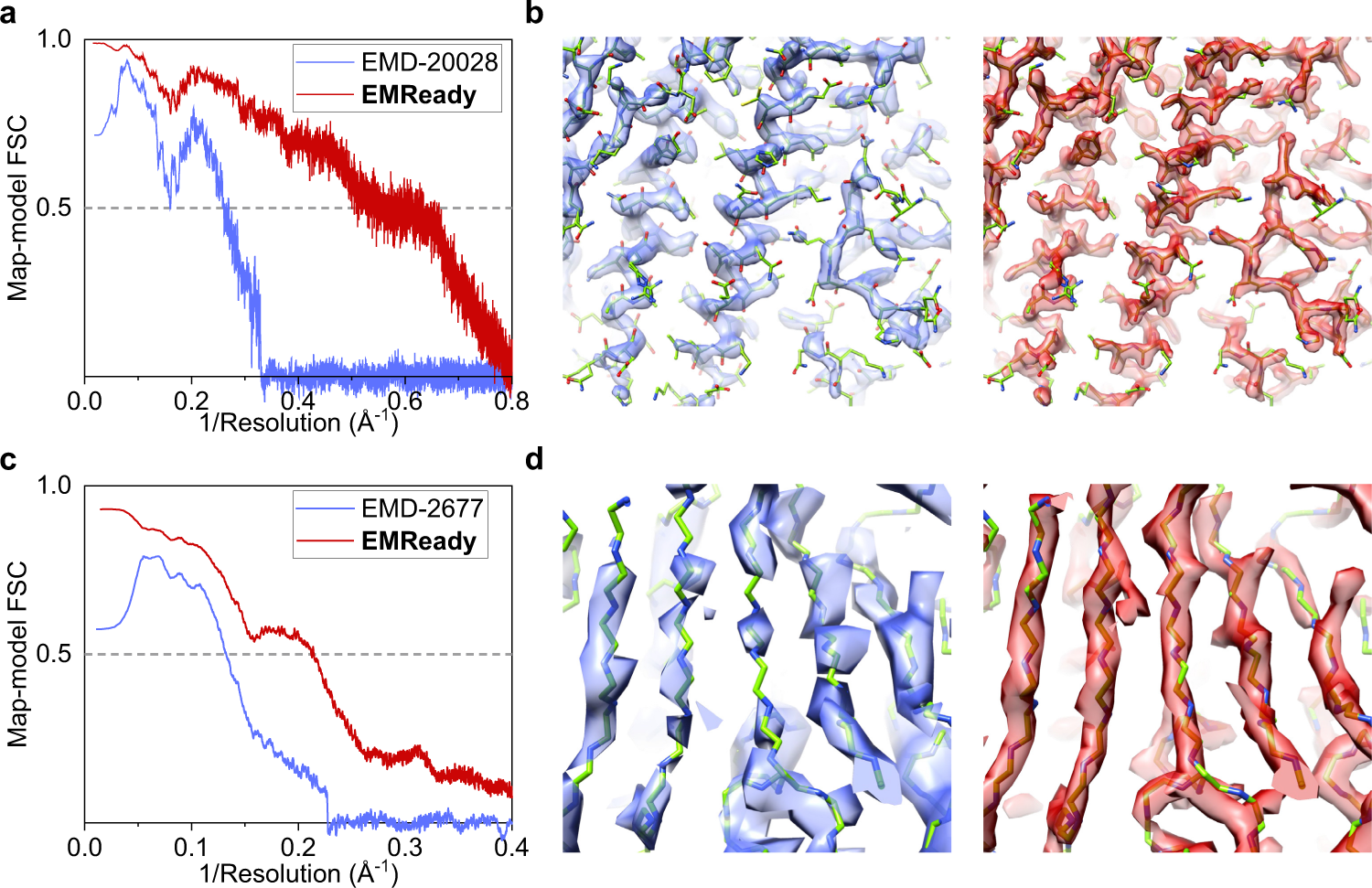
针对这一问题,黄胜友教授课题组提出了一种基于深度学习的三维密度图后处理算法-EMReady,通过同时进行局部和非局部的深度学习来提高密度图的可解释性。EMReady采用三维SCUNet网络架构,结合了用于局部学习的传统残差卷模块和用于非局部学习的移动窗口自注意力模块,并将这两种模块整合到了多尺度的UNet中,以进一步增强其局部和非局部学习的能力。移动窗口自注意力是一种高效的自注意力机制,结合了多个不重叠的窗口内的局部自注意力和通过移动窗口分区实现的非局部跨窗口连接。EMReady的局部和非局部学习能力不仅通过网络架构实现,还在训练过程中通过同时最小化局部的平滑L1距离和最大化非局部的结构相似性来实现,可以有效地避免模型产生过拟合。通过在多个测试集上对EMReady进行了广泛的评估,并与其他密度图后处理算法进行了比较。结果表明,EMReady显著优于其他后处理算法,不仅能够在各种质量评估指标方面显著提高密度图的质量,而且还可以使从头建模算法从密度图中构建出更高质量的蛋白质结构模型。
黄胜友教授课题组多年来一直致力于生物大分子结构及其相互作用的计算与预测研究,在蛋白质-蛋白质、蛋白质-多肽、蛋白质-小分子和蛋白质-核酸相互作用及其复合物结构预测方面做了许多重要的工作,开发了一系列算法、软件和计算平台,并在Nature Protocols、Nature Communications、Journal of the American Chemical Society、Proceedings of National Academy of Sciences、Nucleic Acids Research、Bioinformatics、Drug Discovery Today等国际著名期刊发表多篇论文。
该项研究工作得到了国家自然科学基金(32161133002,62072199)和学校人才引进基金的资助。
论文链接:https://www.nature.com/articles/s41467-023-39031-1

